Projects of Biomolecular Chemistry group
- Project 1
- Project 2
- Project 3
- Project 4
- Project 5
The structure of the 2.6 MDa Saccharomyces cerevisiae FAS I
Gipson, Preeti; Mills, Deryck J.; Wouts, Remco; Grininger, Martin; Vonck, Janet; Kühlbrandt, Werner. (2010) Direct structural insight into the substrate-shuttling mechanism of yeast fatty acid synthase by electron cryomicroscopy. Proc Natl Acad Sci USA, 107, 9164–9169.
and
Johansson, Patrik; Wiltschi, Birgit; Kumari, Preeti; Kessler, Brigitte; Vonrhein, Clemens; Vonck, Janet; Oesterhelt, Dieter; Grininger, Martin. (2008) Inhibition of the fungal fatty acid synthase type I multienzyme complex. Proc Natl Acad Sci USA, 105, 12803–12808.
The 2.6 MDa fungal fatty acid synthases type I (FAS I) consists of two dome-shaped reaction chambers formed by six β-chains, each 230 kDa in molecular weight, and an equatorial wheel-like structure made-up by six α-chains, each 210 kDa. The catalytic domains are interspersed by a number of scaffolding domains, which allow the formation of the complex barrel-shaped architecture, in which the catalytic domains are directing towards the hollow reaction chambers.
In FAS I, the domain ACP has a key role. It takes up substrates and carriers them from one enzymatic domain to the next. S. cerevisiae FAS I proved to be an excellent system for studying substrate shuttling in more detail. In collaboration with the Max-Planck-Institute of Biophysics, we were able to give direct insights in substrate-shuttling by finding ACP docked at active sites.
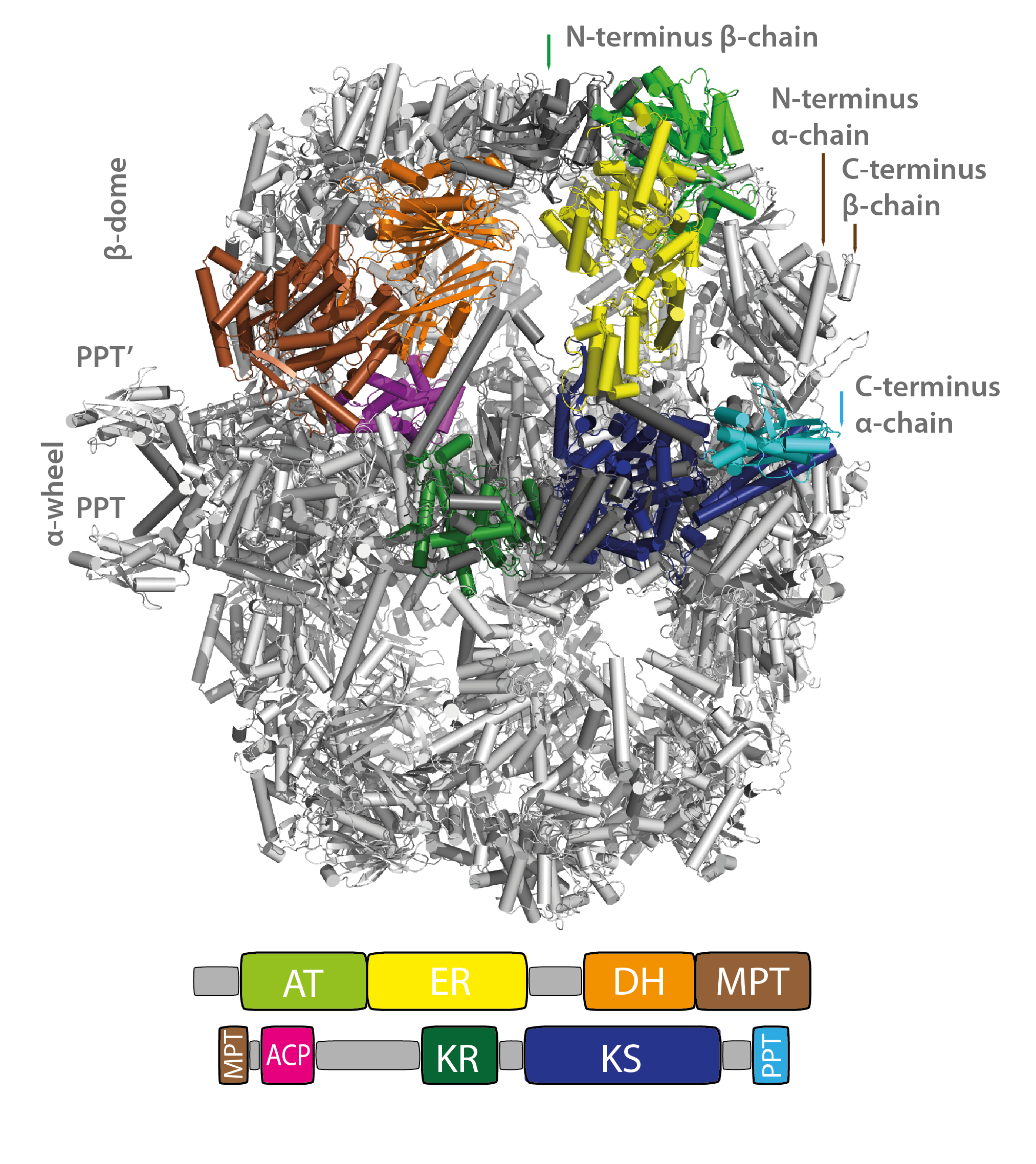
Figure. Structure and domain organization of the fungal FAS I. The S. cerevisiae FAS I is composed of α- and β-chains which assemble to a highly symmetric, 2.6 MDa large heterododecameric (α6β6) complex. Single-chains of the FAS I are colored according to the attached domain overview. Structural elements are indicated. An interesting feature is that fungal FAS I carries phosphopantetheine transferase (PPT) domains as part of their polypeptide chains. PPT is responsible for the initial activation of the FAS by post-translational modification of the ACP domain. PPT domains are attached at the outside.
Further information: The ACP mode of action can be compared to a robotic arm, which supplies substrates to productive sites. However, this comparison is also misleading, as a robotic arm gains productivity by the programming of its pathway, while ACP movement is stochastic. In this manner, the benefit for the FAS I systems is, essentially, to keep the intermediates within the catalytic compartment.
Interestingly, recent structural data refined the mechanistic view onto substrate shuttling, as it emerged to be well-tuned in the interplay of the mobile ACP with the FAS I catalytic body.
The structure of the 1.9 MDa Mycobacterium tuberculosis FAS I
Ciccarelli, Luciano; Connell, Sean; Enderle, Mathias; Mills, Deryck J.; Vonck, Janet; Grininger, Martin. (2013) Structure and conformational variability of the Mycobacterium tuberculosis fatty acid synthase multienzyme complex, Structure, 21, 1251-1257.
The bacterial fatty acid synthase type I (FAS I) is a 2.0 MDa large multienzymatic molecular machine. Although considerably reduced in molecular weight compared to fungal FAS I, the evolutionary relationship to fungal FAS I was evident from sequence comparison. In a recent work on the electron microscopic characterization, we revealed that, although overall homologous to rigid fungal FAS I, the bacterial FAS I is of high conformational variability allowing dynamic domain reorganizations during fatty acid synthesis.
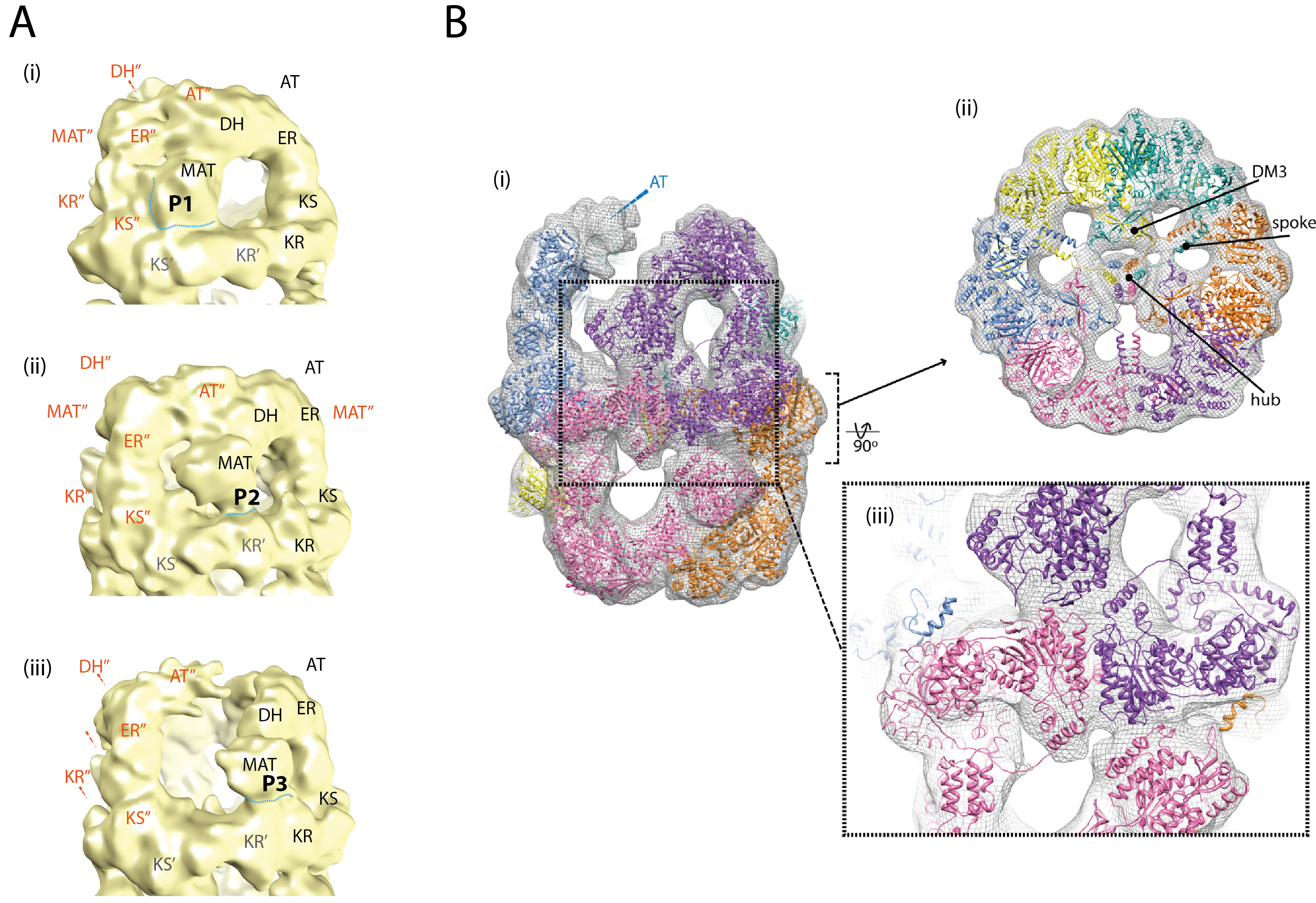
Figure. Domain arrangements in M. tuberculosis FAS I and molecular model of state P3. (A) MAT positions P1 (i), P2 (ii) and P3 (iii). In EM maps 1-3, three states could be identified with characteristic positioning of domains. As in this states MAT is displaced largest, the states are denoted P1, P2 and P3 according to MAT positions. The domain labels KS, KR, DH, ER, AT and MAT in each chain are distinctly colored and denoted with a superscript. (B) Homology model of M. smegmatis FAS I fitted into M. tuberculosis FAS I map 1 with state P3 in the upper dome. Figure (i) shows the FAS I complex from the side, while the upper right figure shows the central section (central wheel) from the top. At the broken interface, the density of the AT domain of the neighboring chain (blue) is fragmented probably due to an increased flexibility induced by disruption of stabilizing interactions formed with the shifted DH/MAT domains. The uncertainty in the placement of the AT domain prompted to omit it from the model. Figure (ii) shows the overall conservation of features between the M. tuberculosis and the M. smegmatis FAS I on the example of the central wheel. Important structural features, such as DM3, spoke and the central hub are labeled. In (iii), a side view of the FAS complex is shown, focusing on the opening chain (purple) and its interface with the adjacent chain (pink) forming the opposite dome.
Further information: In addition to the broad impact of our work on the understanding of bacterial FAS I systems, the presented data are also of specific importance. M. tuberculosis is the causative agent of tuberculosis (TB). Its FAS I is responsible for de novo fatty acid synthesis, forming precursors for mycolic acids that build up the impermeable, wax-like cell wall and make M. tuberculosis impervious to medical drugs. The relevance of M. tuberculosis FAS I as drug target is documented by pyrazinamide, which has been identified as specifically targeting M. tuberculosis FAS I and is used in current antibiotic therapy.
We consider our data as highly relevant for anti-tuberculosis initiatives. We demonstrated that M. tuberculosis FAS I can be recombinantly expressed in Escherichia coli, which will facilitate future functional and structural investigations on this highly demanded drug target. Moreover, our data potentially contribute in establishing M. tuberculosis FAS I as a target for rational drug design.
Assembly of MDa-sized FAS oligomeric structures
Johansson, Patrik; Mulinacci, Barbara; Koestler, Caecilia; Vollrath, Ronnald; Oesterhelt, Dieter; Grininger, Martin. (2009) Multimeric options for the auto-activation of the Saccharomyces cerevisiae FAS type I megasynthase. Structure, 17, 1063–1077.
S. cerevisiae fatty acid synthase type I (FAS I) is well studied; however, the assembly pathway of the heterododecamer is essentially unknown. Recent X-ray crystallographic studies revealed a functional conundrum. The ACP domain has to be post-translationally modified by a phosphopantetheine transferase (PPT) domain to drive fatty acid synthesis. However, in the fatty acid synthesis competent α6β6 FAS I particle, ACP and PPT are spatially separated so that post-translational modification cannot occur. Consequently, for full catalytic activity, the assembly of S. cerevisae FAS I from its subunits has to include a defined intermediate state where ACP and PPT can interact. Recently, we were able to show that PPT has to oligomerize to a dimer or trimer to be catalytically active, which constraints the possible conformations of a pre-complex.
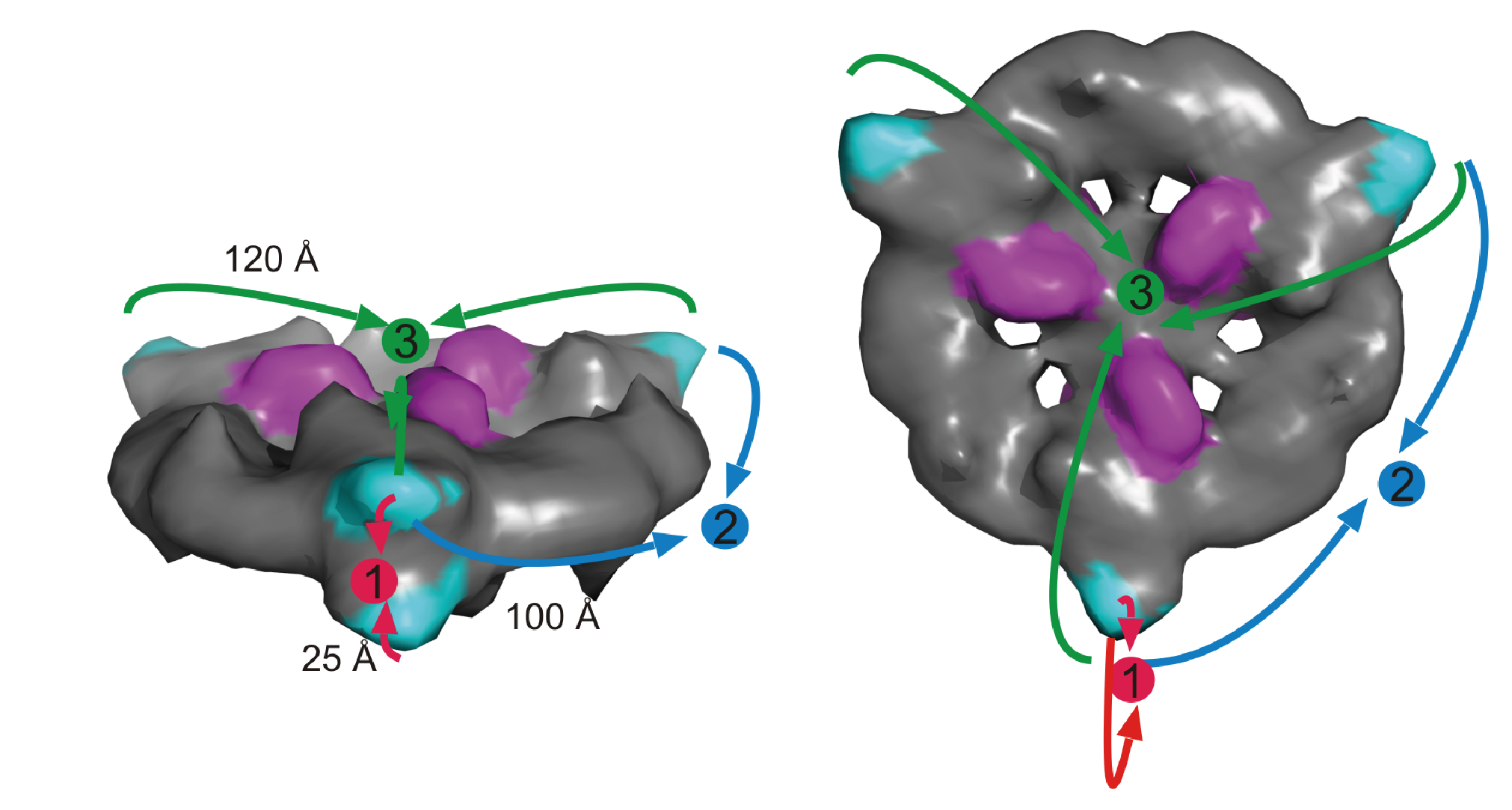
Figure. ACP-PPT interaction within a α6β6 FAS I particle. Besides the spatial separation of ACP and PPT in fungal FAS I, the PPT and ACP cannot interact owing to spatial restraints. This figure shows the separation of ACP (magenta) and PPT (cyan). For clarity, a (physiologically non-relevant) α6 wheel substructure is shown. A sphere with a probe radius of 18 Å was used for surface calculation.
Further information: Under the assumption of conserved assembly pathways among several types of fungal FAS I, the interaction of β- and α-chain might be an early event in fungal FAS I assembly. Considering an early interaction of chains and comforting the dimeric character of the domains KS and KR as well as of scaffolding elements, a pre-complex could have a α2β2 nature.
Engineering the structural properties of dodecin
Yu, Yaming; Heidel, Björn; Parapugna, Tamara; Wenderhold-Reeb, Sabine; Song, Bo, Schönherr, Holger; Grininger, Martin; Nöll, Gilbert. (2013) The flavoprotein dodecin as redox probe for ET through DNA, Angew. Chem. Int. Ed. Eng., 52, 4950-4953.
and
Grininger, Martin; Nöll, Gilbert, Trawöger, Sibylle; Sinner, Eva-Kathrin; Oesterhelt, Dieter. (2008) Electrochemical switching of the flavoprotein dodecin at gold surfaces modified by flavin-DNA hybrid linkers. Biointerphases, 3, 51–58.
The sensitivity of dodecin towards the redox-state of the bound flavin represents one of its key properties. While dodecin binds oxidized flavin systems with high affinities, reduced compounds cannot be adopted. On this basis, we have engineered dodecin variants to bind flavins with low substrate specificity. We have employed them as a redox-sensitive shuttle, which can be loaded with flavin-anchored compounds, and use them as a redox-sensitive load of surface anchored flavins. For the latter, it requires a surface tethered flavin (flavin/molecular wire hybrid), which enables electron transfer from the surface (electrode) to the flavin head group (isoalloxazine ring).
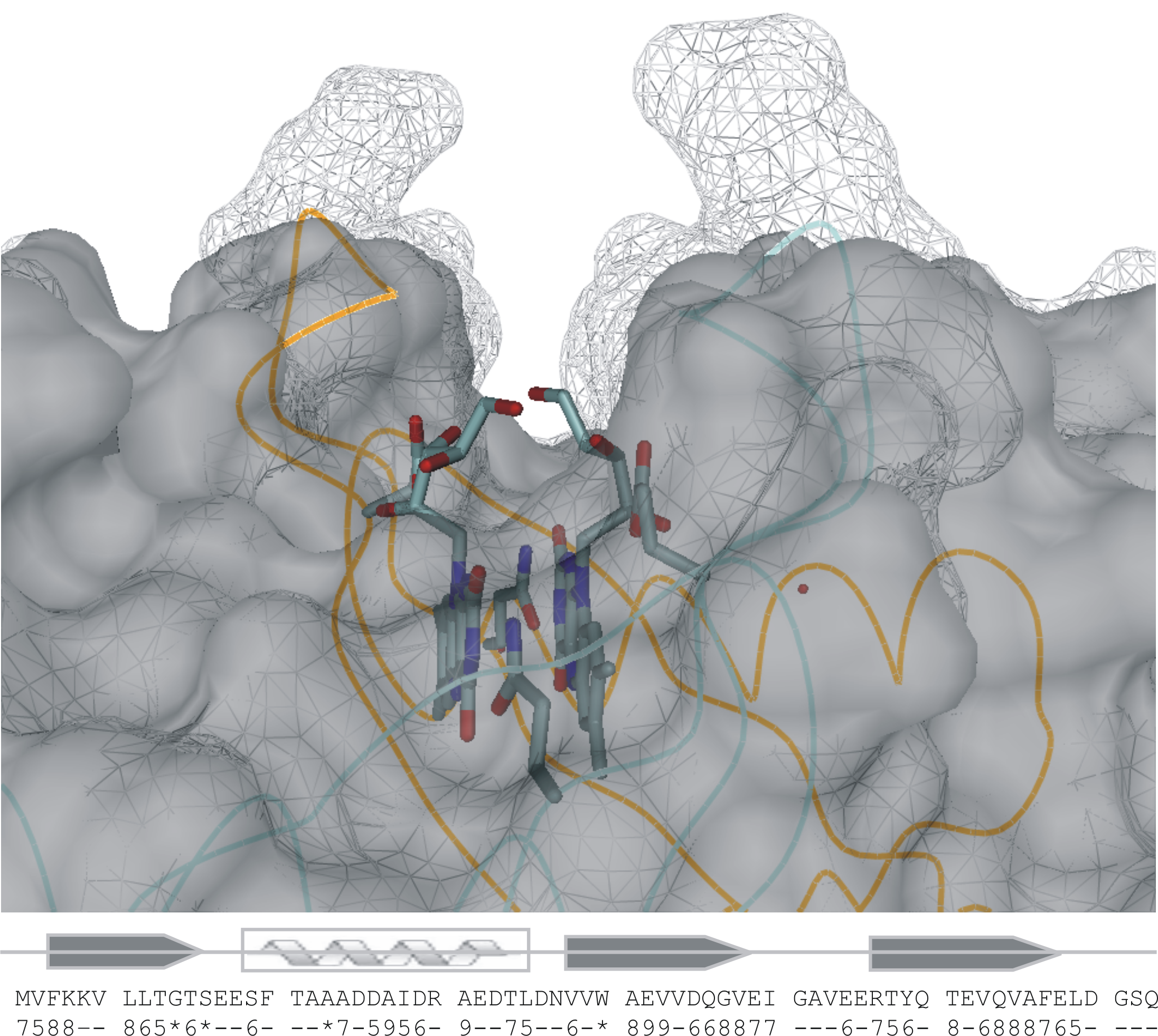
Figure. Superposition of the binding pockets of wildtype and tE-mutated dodecin. The binding pocket of the wildtype dodecin (mesh representation) and the engineered dodecin tE variant (solid), which is tolerant in ligand binding, are shown in a surface representation mode. The Cα-chains of two dodecin wildtype monomers are traced by orange and grey lines, respectively. The dodecin secondary structure and the primary sequence are attached, with the conservation of the primary sequence per 10 dodecin homologs. A star indicates full conservation; lines indicate conservations below 50% (conservation code 5).
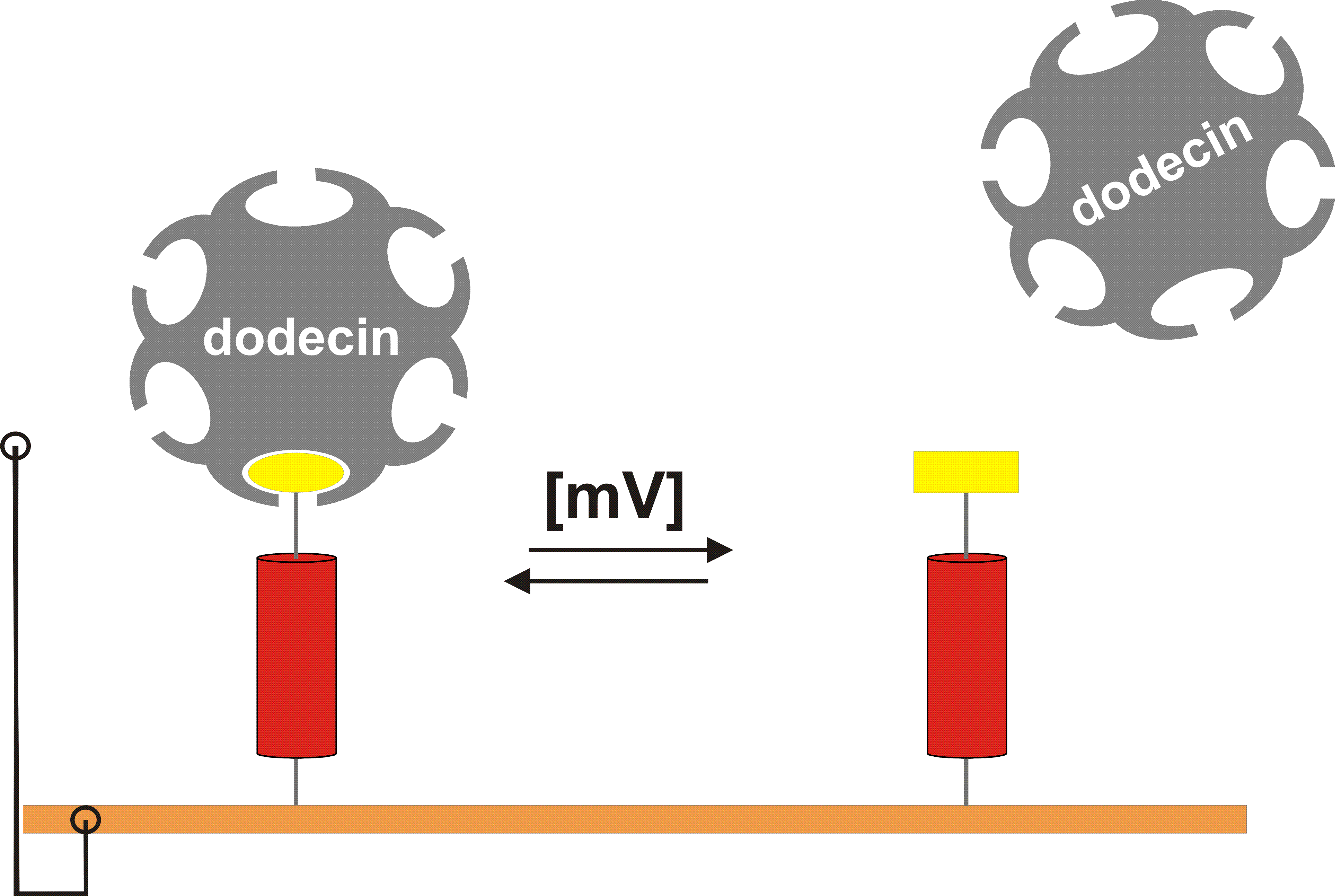 Figure. Electrochemically switching of protein states. Flavin/molecular wire ligand hybrids are linked to electrode surfaces. They are switched between oxidized (left; flavin abstracted as ellipse) and reduced state (right, rectangle) by the surface potential. Driving force for a changed binding behavior are different electronic and conformational properties of flavins in the oxidized vs. the reduced state.
Figure. Electrochemically switching of protein states. Flavin/molecular wire ligand hybrids are linked to electrode surfaces. They are switched between oxidized (left; flavin abstracted as ellipse) and reduced state (right, rectangle) by the surface potential. Driving force for a changed binding behavior are different electronic and conformational properties of flavins in the oxidized vs. the reduced state.
Further Information: Dodecin is a flavin binding protein, which was first found in the archaeal organism Halobacterium salinarum. It forms a dodecameric, hollow-spherical structure of a diameter of about 7 nm. A single protein consists of 68 amino acid residues, representing the smallest flavoprotein known to date. Sequence alignments reveal that the dodecin gene is widely distributed within bacteria and haloarchaea. Interestingly, a significant number of bacteria bearing dodecins are human pathogens and exposed to an increased oxygen concentration or radical stress through host defense.
Dodecin exhibits a new fold of simple β1-α-β2-β3-β4-topology, comprising an α-helix partly enwrapped by a three stranded antiparallel β-sheet. Of particular structural interest is the aromatic tetrade, which contains two π-stacked flavins embedded by two tryptophans. This binding motif is unique, as the dodecin binding pocket is not designed complementary to the chemistry of a single ligand, but to the chemistry of ligand dimers.
 Figure. The structure of dodecin. (A) View on the highly symmetrical dodecin particle, along a three-fold (view 1), and a two-fold axis (view 2). A β-sheet network forms the hollow-sphere and the α-helices are exposed to the bulk water. (B) Detailed view on the aromatic tetrade, consisting of the π-staggered indole groups of tryptophan Trp36 and the isoalloxazine-ring of flavins. In the right figure, dodecin is represented by its surface and the aromatic tetrade is reduced to the C2-related part for clarity. The aromatic ring systems are placed in a distance of about 3.3 Å. In this co-planar arrangement, the isoalloxazine-rings contact each other with the ribityl-chains pointing in the same direction towards the outer surface of the protein. The dominant polar interactions to the aromatic system are two hydrogen bonds of the amide side chain of Gln55 (not shown). Glu45 fixes the ribityl chain by hydrogen bonding to hydroxyl groups.
Figure. The structure of dodecin. (A) View on the highly symmetrical dodecin particle, along a three-fold (view 1), and a two-fold axis (view 2). A β-sheet network forms the hollow-sphere and the α-helices are exposed to the bulk water. (B) Detailed view on the aromatic tetrade, consisting of the π-staggered indole groups of tryptophan Trp36 and the isoalloxazine-ring of flavins. In the right figure, dodecin is represented by its surface and the aromatic tetrade is reduced to the C2-related part for clarity. The aromatic ring systems are placed in a distance of about 3.3 Å. In this co-planar arrangement, the isoalloxazine-rings contact each other with the ribityl-chains pointing in the same direction towards the outer surface of the protein. The dominant polar interactions to the aromatic system are two hydrogen bonds of the amide side chain of Gln55 (not shown). Glu45 fixes the ribityl chain by hydrogen bonding to hydroxyl groups.
Reaction control in dodecin
Staudt, Heike; Hoesl, Michael Georg; Dreuw, Andreas; Serdjukow, Sascha; Oesterhelt, Dieter; Budisa, Nediljko; Wachtveitl, Josef; Grininger, Martin. (2013) Directed manipulation of a flavoprotein photocycle. Angew Chem Int Ed Eng., 52, 8463-8466. paper awarded as “hot paper”
For manipulating and controlling dodecin reaction specificity, we set out to modify the ionization potential of the tryptophan W36 adjacent to the riboflavin ligand. Upon illumination, W36 donates electrons to a light-activated riboflavin, which then quenches riboflavin active states. A changed ionization potential should therefore interfere in the electron transfer rate. We calculated ionization potential for W36 derivatives, modified at the 4-position, and then incorporated selected W36 derivatives into the protein by bioorganic methods. Electron transfer rates behaved as predicted, and were faster or slower in accordance to the ionization potential. The dodecin variants, carrying the non-native W36 derivatives, did not show any structural perturbations, as tested by X-ray crystallographic studies.
Thus, we introduced chemical modifications in a non-invasive manner to receive isomorphous proteins with functional characteristics changed as predicted by quantum chemical methods.
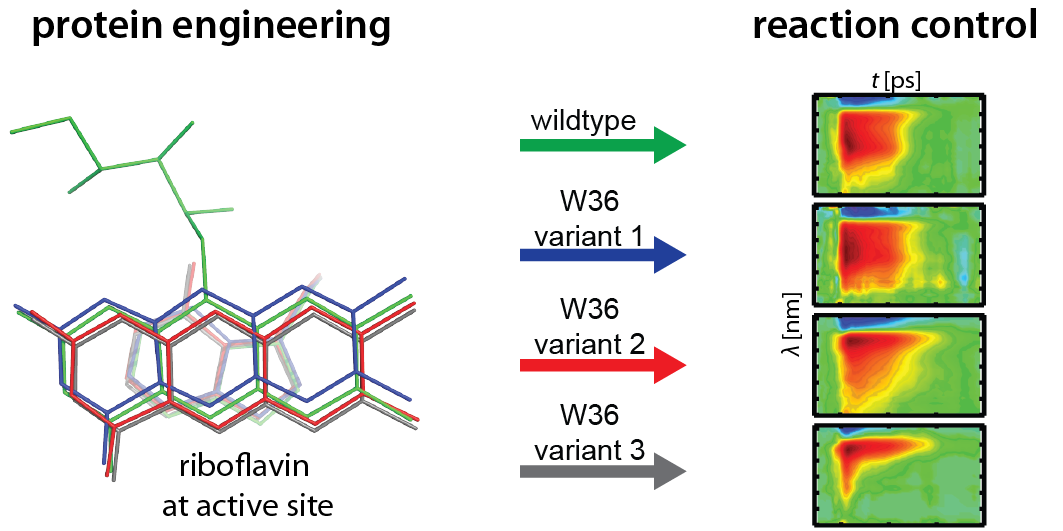
Figure. Rational design of dodecin for manipulation of the dodecin photocycle. Guided by quantum chemical calculations and high-resolution X-ray crystallography, we have exchanged the electron donating tryptophan by non-native derivatives, to generate a series of dodecins, which are modulated in electronic properties but virtually unchanged in structure. We finally used ultra-fast spectroscopy to show that the non-native dodecins are unaffected in the reaction cascade of the dodecin photocycle, but selectively modulated in the lifetimes of their intermediates.
Further Information: We are generally interested in the modification of proteins to optimize protein properties for beneficial applications or for learning about protein functions. Engineering can address substrate specificity, or reaction specificity. Substrate specificity is an important parameter for protein function. It is a carefully modulated intrinsic property of proteins, for example in case of DNA-polymerases, allowing the reaction with various 3-desoxy-nucleotides, but not with other nucleotides or chemically similar cofactors. Dependent on the specific protein, substrate specificity can be easily engineered, as also described with dodecin (see above). In contrast, reaction specificity, which is the specific chemical transformation performed within a catalytic site, is difficult to engineer. It is realized by accurately positioned chemical functions within an active site, and generally transforming chemical functions in a defined and invariable manner. For dodecin, we have achieved to interfere in reaction specificity and have manipulated the light cycle in a rational manner.